Clinical Description
CASK disorders are more commonly reported in females and include a spectrum of phenotypes that differs in females and males:
Females typically have moderate-to-severe intellectual disability and in most individuals, progressive microcephaly with pontine and cerebellar hypoplasia (MICPCH). Possible findings are ophthalmologic anomalies and sensorineural hearing loss. Females who are relatives of males with the
X-linked intellectual disability (XLID) ± nystagmus
phenotype may rarely present with a mild-to-severe intellectual disability phenotype.
In males the spectrum is broad, ranging from severe (intellectual disability and MICPCH, or early-infantile epileptic encephalopathy [Ohtahara syndrome, West syndrome, or early myoclonic epilepsy]) to mild (XLID ± nystagmus and additional clinical features) [
Moog et al 2015].
To date, 130 individuals (45 males and 85 females) have been identified with a pathogenic variant in CASK [Moog et al 2011, Burglen et al 2012, Hayashi et al 2012, Takanashi et al 2012, Moog et al 2015, Dunn et al 2017, Hayashi et al 2017, Muthusamy et al 2017, Cristofoli et al 2018, Rama Devi et al 2019]. The following description of the phenotypic features associated with this condition is based on these reports.
Females
A total of 85 females with MICPCH have been reported to date, the eldest of whom is age 25 years. The following information about the natural history is based on the recent reviews of Moog et al [2011], Burglen et al [2012], Hayashi et al [2012], and Takanashi et al [2012] unless otherwise noted.
Microcephaly with Pontine and Cerebellar Hypoplasia (MICPCH)
Head circumference. At birth the occipital frontal circumference (OFC) is in the normal or low-normal range in approximately two thirds of affected females; the others show microcephaly (OFC < -2 SD). Microcephaly invariably becomes severe (OFC -3.5 to -10 SD) during the first year, and usually during the first four months of life.
Developmental delay / intellectual disability (DD/ID). Affected females acquire head control and make eye contact in the range of two to 24 months. Most affected females are able to sit independently between seven and 36 months; only 20%-25% attain the ability to walk (between 18 and 72 months).
Language is nearly absent in most; some utter words. One individual could say two-word sentences. Intellectual development is severely impaired in nearly all affected females, with a few showing moderate ID.
The behavioral phenotype may include sleep disturbances, hand stereotypies, and self biting.
Neurologic features include (axial) hypotonia, hypertonia of the extremities (possibly progressing to spasticity), and dystonia or other movement disorders. Seizures of various types are observed in about 40%; onset is between birth and age ten years.
The severity of the pontocerebellar hypoplasia observed on MRI is not of prognostic value [Moog et al 2011].
MRI findings
Normal- or low normal-sized corpus callosum with low cerebrum / corpus callosum ratio [
Takanashi et al 2010]
Associated MRI finding: mildly reduced number and complexity of gyri in the frontal region of the cerebral cortex and mild dilatation of the lateral ventricles [
Moog et al 2011]
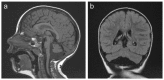
MRI of the brain of a girl age 2.5 years with MICPCH and a heterozygous CASK pathogenic variant a. Sagittal image showing mild pontocerebellar hypoplasia with sparing of pontine bulging. The corpus callosum is normal.
Other findings
Scoliosis is frequently observed.
Various ophthalmologic findings can be observed, in particular optic nerve hypoplasia, retinopathy, nystagmus, and strabismus [
LaConte et al 2019].
Congenital visceral anomalies (e.g., renal/urologic or cardiac anomalies) are rarely seen; no particular anomaly occurs recurrently.
Recent reviews suggest a facial
phenotype consisting of well-drawn arched eyebrows, a broad nasal bridge and tip, small or short nose, long philtrum or protruding maxilla, small chin, and large ears.
Mortality in affected females has not been reported.
X-Linked Intellectual Disability (XLID) ± Nystagmus
Clinical findings in the majority of heterozygotes (typically identified as relatives of more severely affected males):
Normal intelligence; mild-to-severe ID in some females only
Normal-to-mild ocular findings including
congenital nystagmus and strabismus
No additional neurologic signs besides mild tremor or absence seizures
MRI finding: normal or mainly unknown
Males
A total of 45 males from birth to age 59 years with a pathogenic CASK variant have been described [Moog et al 2015, Dunn et al 2017, Hayashi et al 2017, Muthusamy et al 2017, Rama Devi et al 2019].
The phenotype in males represents a clinical continuum from the severe to the mild end of the spectrum and can be classified into three phenotypic groups [Moog et al 2015].
MICPCH with Severe Epileptic Encephalopathy
Head circumference. At birth, the OFC was (low) normal in half of the individuals. The other half had primary microcephaly (OFC <-2 SD). Mild-to-severe postnatal microcephaly evolved rapidly during the first months (OFC -2.7 to -9 SD).
DD/ID. All affected males had severe-to-profound DD or no development at all.
Neurologic features include early and intractable seizures (Ohtahara syndrome [Saitsu et al 2012], West syndrome [Takanashi et al 2012], myoclonic epilepsy [Nakamura et al 2014]), burst suppression and spasms [Moog et al 2015], and hyperkinesia [Rama Devi et al 2019].
MRI findings
Typically severe diffuse pontocerebellar hypoplasia
Simplified gyri, cortical atrophy, and hypomyelination may be also observed.
Other findings
Mortality. Males with this phenotype may have perinatal or early lethality. One affected male died at age two months [Rama Devi et al 2019], one at seven months, and another at 21 months [Moog et al 2015].
MICPCH with Severe Developmental Disorder
MICPCH in combination with a severe developmental disorder but without severe epilepsy has been reported in six males. The phenotype of male individuals in this group is comparable to MICPCH in females [Moog et al 2015, Hayashi et al 2017]:
Head circumference. Postnatal microcephaly
DD/ID. Severe
Neurologic features. Mild ataxia reported in one male, dystonia/dyskinesia in another male. No seizures.
MRI findings. Variable degree of diffuse pontocerebellar hypoplasia
Other findings. Nystagmus
Mortality. One affected male died at age two weeks.
X-Linked Intellectual Disability (XLID) ± Nystagmus
Mild-to-severe XLID with or without nystagmus and/or other anomalies have been reported in a total of 29 males [Moog et al 2015, Dunn et al 2017, Hayashi et al 2017].
Brain MRI has been reported in a minority of individuals only and did not show pontocerebellar hypoplasia.
Other findings include microcephaly, hypotonia, autism spectrum disorder, behavioral problems, tremor and unsteady gait, sensorineural hearing loss, feeding difficulties, constipation, short stature, cryptorchidism, and gastrointestinal and gastroesophageal complications.
Genotype-Phenotype Correlations
In females, microcephaly with pontine and cerebellar hypoplasia (MICPCH) is typically associated with heterozygous CASK pathogenic loss-of-function variants [Moog et al 2011, Burglen et al 2012, Hayashi et al 2012, Takanashi et al 2012, Hayashi et al 2017]. The X-linked intellectual disability (XLID) with or without nystagmus phenotype in females is typically associated with CASK hypomorphic pathogenic variants.
In males, the three clinically distinguishable groups are associated with different classes of pathogenic CASK variants [Moog et al 2015]:
In males with MICPCH with severe epileptic encephalopathy, the most severe
phenotype, the majority of
CASK pathogenic variants are
germline loss-of-function alterations.
In the group with MICPCH, males are somatic mosaics of a
CASK loss-of-function variant or carry partly penetrant variants in the
hemizygous state.
The largest group of males with XLID with or without nystagmus typically have
CASK hypomorphic pathogenic variants, including
missense and splice variants [
Moog et al 2015].